RESULTS
OF NOVEL STRATEGIES FOR TREATMENT OF WILMS’ TUMOR
(
Download pdf )
SILVIO TUCCI JR, ADAUTO J. COLOGNA, HAYLTON J. SUAID, ELVIS T. VALERA, LUIS F. TIRAPELLI, EDSON L. PASCHOALIN, ANTONIO C. MARTINS
Division of Urology, Ribeirao Preto Medical School, University of Sao Paulo, Ribeirao Preto, Sao Paulo, Brazil
ABSTRACT
Objective:
To evaluate treatment outcomes in Wilms’ tumor (WT).
Materials and Methods: We studied 53 children
with median age of 2 years with WT, stages I-19, II-14, III-12, IV-6 and
V-2. Treatment consisted of surgical excision plus adjuvant (40 children)
or neoadjuvant and adjuvant chemotherapy (unresectable tumor, n = 8, or
caval tumor extension, n = 5). Chemotherapy and radiotherapy followed
protocols of Brazilian Wilms’ Tumor Study Group excepting 16 cases
with stage I disease that received a short duration postoperative treatment
with vincristine (VCR - 11 doses) and dactinomycin (AMD - 4 doses). Relapsed
WT was treated with multiagent regimens including cisplatin/carboplatin,
cyclophosphamide, ifosfamide and etoposide. One patient with resistant
relapsed WT was treated by high-dose conditioning chemotherapy with stem
cell rescue.
Results: Overall and disease-free survival
rates at 5 years were respectively 88.2 ± 5.0% and 76.7 ±
6.6%. Short duration therapy for stage I tumor showed a disease-free survival
rate of 100% in a median time of 101 months (range 14 to 248 months).
Overall and disease-free survival of 10 patients with recurrent WT at
5 years was 42.8%. The child treated with high-dose chemotherapy plus
stem cell transplant is alive without evidence of disease 84 months from
relapse.
Conclusion: The postoperative chemotherapy
in stage I disease can be reduced without compromising the cure rate.
The treatment of unfavorable stage III and IV disease or relapsed tumor
remains a challenge.
Key
words: Wilms’ tumor; surgery; chemotherapy; relapse
Int Braz J Urol. 2007; 33: 195-203
INTRODUCTION
The
multidisciplinary management of Wilms´ tumor (WT) led to a striking
improvement of patient outcome in the last decades. Now, an increasing
consideration is given to determine the minimal therapy needed to cure
low-risk tumors (1,2). Recent WT trials of the International Society of
Pediatric Oncology (SIOP) and United Kingdom Children’s Cancer Study
Group (UKCCSG) has shown that chemotherapy with vincristine (VCR) or a
combination of VCR and dactinomycin (AMD) for stage I favorable histology
tumor can be reduced without compromising survival rates, but there is
no agreement on the matter (1-4). At the same time, researchers are looking
for novel strategies such as treatment intensification for children with
high-risk tumors (5). The use of modern intensive regimens including doxorubicin
(ADR), cyclophosphamide (CTX), ifosfamide (IFO), carboplatin (CP) and
etoposide (ETP) were reported to improve survival rates for patients with
favorable relapsed WT from less than 30% to 50-55% (5-7). However, children
with unfavorable relapsed WT have a high risk of treatment failure (1,7).
Few reports on high-dose chemotherapy with autologous stem-cell rescue
show variable disease-free survival rates (33 to 60%) and it is not clear
whether this approach offers any advantages over conventional second line
therapies (1,5,8-10).
The aim of this study is to analyze the
outcomes in children with WT treated in a single center with special attention
to stage I disease and to patients with high-risk recurrent disease.
MATERIALS AND METHODS
We
reviewed the records of 53 children (28 males and 25 females) with WT
who were treated at our institution between January 1980 and December
2004. Median patient age was 2 years (range < 1 to 14). Inclusion criteria
were adequate clinical and pathological data, and a follow-up of 1 year
or more, except for those who died of the disease. Two patients were excluded
from the study because 1 died just after arrival, and another one that
died in the 1st postoperative day of a nephrectomy.
Tumor stage and histological subtype (Table-1)
were defined according to the National Wilms’ Tumor Study Group
(NWTSG) (1,11).

Surgical treatment for unilateral disease
consisted of transperitoneal radical nephrectomy. One patient with stage
V tumor was submitted to bilateral biopsies followed by chemotherapy and
then to total unilateral nephrectomy and partial contralateral nephrectomy.
Other child with stage V in whom a very small nodule in the left kidney
was not noticed at presentation was treated initially by unilateral right
nephrectomy followed by chemotherapy and afterward by partial left nephrectomy.
Regional lymph node sampling was obtained in all patients and an associated
adrenalectomy was performed in 16 cases, enterectomy in 2 and caval thrombectomy
in 5. Forty patients were treated with adjuvant chemotherapy only. Thirteen
children with tumors initially deemed unresectable or with intracaval
extension received neoadjuvant and adjuvant therapy as follows: stages
I - 3, II - 1, III - 4, IV - 3 and V - 1.
With exception of patients with stage I
tumor, the routine adjuvant chemotherapy and radiotherapy followed the
recommendations of the Brazilian Wilms’ Tumor Study Group (12).
Most patients with stage I tumor (16/19) received a shorter postoperative
treatment with 11 doses of VCR (1.5 mg/m2, weeks 2-11 and 16) and 4 doses
of AMD (15-60mg/kg, weeks 1, 6, 11 and 16).
Relapsed WT was treated with multiagent
salvage regimens including CIS, CP, CTX, IFO and ETP (6,7). In addition,
abdomen radiotherapy was used in 6 cases (10.5 to 30 Gy) and lung radiotherapy
in 3 (12 to 15 Gy). One patient with WT (metastasis in liver and lungs)
resistant to salvage regimen (6 courses using a combination of CIS, ETP
and IFO) was treated by a lobectomy for resection of 2 residual lung metastasis
(the nodule in the liver was unsuitable for surgical resection), followed
by radiotherapy (12 Gy in both lungs and 12 Gy in the liver), and high-dose
conditioning chemotherapy (high-dose chemotherapy - ETP 170 mg/m2, melphalan
140 mg/m2 and CP 600 mg/m2) with stem cell rescue (2.7 x106 cells/kg)
(9). Autologous CD34 cells were harvested by aphaeresis 45 and 30 days
before conditioning therapy after a 5 days course of GM-CSF (10mg/kg/day).
The median follow-up was 58 months (range
7 to 274). Survival rates were calculated by the Kaplan-Meier method.
Comparisons between groups of patients were made using log rank univariate
analysis and Cox regression multivariate analysis. The statistical analysis
was performed using Stataâ 6.0 software. P values less than 0.05
were considered significant.
RESULTS
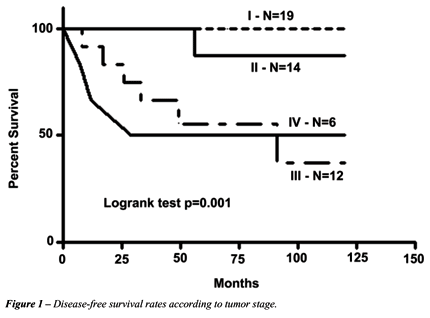
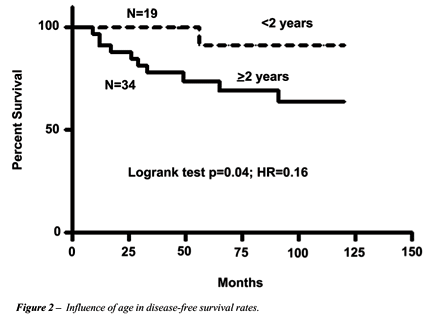
Overall
and disease-free survival rates were displayed in Figures-1 and 2 as well
as in Tables-2, 3 and 4. Multivariate analysis shows that influence of
tumor stage is more relevant (p = 0.03; HR = 1.56) than children age (p
= 0.05; HR = 0.39) on disease-free survival rates.



One patient out of 3 with stage I tumor
treated with neodjuvant plus adjuvant chemotherapy died of pneumonia 11
months after nephrectomy. The other 2 are alive disease free 240 and 274
months from surgery. The 16 patients with stage I tumor treated with short
duration chemotherapy showed a disease-free survival rate of 100% in a
median time of 101 months (range 14 to 248 months). Only 1 of these 16
patients developed moderate toxicity represented by neutropenia and infection
(pneumonia) that was treated successfully by dose reduction and antibiotics.
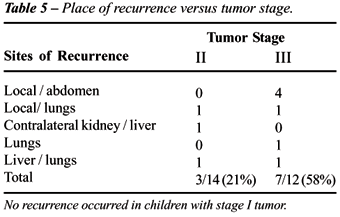
Ten children showed recurrent WT (Table-5)
in a mean time of 13.4 ± 10 months (range 2 to 36). Overall and
disease-free survivals of patients with recurrent WT at 3 and 5 years
were respectively: 83.3% and 66.6%, and 42.8% and 42.8%. Severe drug toxicity
in patients with relapse, treated with salvage chemotherapy, occurred
in 1 patient that developed cardiac insufficiency. The child treated with
high-dose chemotherapy plus stem cell rescue is alive without disease
(hepatic metastasis exhibited complete remission) 84 months from relapse,
but acute drug toxicity due to this attempt was severe: mucositis, vomiting,
diarrhea, seizures, acute pulmonary edema and jaundice. The patient engrafted
to an absolute neutrophil count 500/mL and platelet count > 20/µL,
respectively 18 and 210 days after stem cell transplant.
Only 1/16 adrenal glands excised during
radical nephrectomy had WT metastasis (there was no primary tumor contiguous
involvement). No major perioperative surgical complication occurred in
any children, but 3 cases showed small bowel obstruction 3, 14 and 20
months after the surgery. Only 1 of these 3 children was treated previously
with abdominal radiotherapy. Tumor spill occurred in 5/52 patients: 2
local and 3 diffuse spill. Two children with diffuse spillage exhibited
local relapse.
COMMENTS
Overall
and disease-free survival rates after 3 and 5 years of follow-up are within
the range reported by others (7,12-14).
Our results show that adjuvant short-duration
VCR-AMD chemotherapy is well tolerated and very effective for children
of all ages with stage I Wilms´ tumor. It is worth to stress that
9/16 children so treated were older than 2 years, no one had diffuse anaplasia
and only 1 had focal anaplasia. The SIOP 93-01 trial showed that postoperative
chemotherapy with VCR-AMD for stage I patients with intermediate-risk
and anaplastic WT (submitted to neoadjuvant therapy with the same drugs
for 4 weeks and that showed initial good response) can be shortened to
4 weeks from the standard 18 weeks, while maintaining equivalent disease-free
survival (4). The UKCCSG showed similar results with adjuvant VCR monotherapy
(duration of 10 weeks) for stage I favorable histology tumor in patients
≤ 2 years (2). The NWTSG-5 trial included a therapy arm in which
no adjuvant treatment was given for small stage I WT in children younger
than 2 years, but this arm was closed prematurely when the relapse-free
survival decreased below 90% (15). The few existing reports, including
our data, suggest that treatment reduction for stage I disease is possible.
Meanwhile, the NWTSG still recommend managing patients with stage I disease
with a standard adjuvant chemotherapy regimen (VCR plus AMD) for 18 weeks
(1).
The outcome of our patients with stage II
disease mirrors what was described in the literature while for stages
III and IV tumor it seems worse (3,13,14). The difference observed in
stages III and IV might be casual or a consequence of disparities in sampling.
Our results show that second line chemotherapy
was quite ineffective for treatment of relapsed WT. However, it is relevant
to mention that only 1/10 recurrent tumor in this setting fulfilled the
favorable prognostic factors such as initial stage I or II, previous treatment
with VCR and AMD only, no previous radiotherapy, relapse longer than 6
months after diagnosis and favorable histology (5-7). Although successful
retrieval of recurrent tumor is possible, novel approaches are urgently
needed. In fact, the treatment for resistant relapsed WT remains a challenge
(16). For such patients, in spite of drug toxicity, a salvage attempt
with high-dose chemotherapy associated with autologous stem cell transplant
seems to be justified as seen in 1 patient of this setting as well as
in small series published elsewhere (6,8-10).
The results of transperitoneal radical nephrectomy
with regard to tumor spill (10%), local recurrence (6%) and small bowel
obstruction (6%) are within the range published elsewhere (17-19). The
role of systematic adrenalectomy in treatment of WT deserves further evaluation
because tumor involvement of the gland is not usual as seen in our data.
CONCLUSIONS
The postoperative chemotherapy in stage I disease can be reduced without compromising the cure rate. The treatment of unfavorable stage III and IV disease or relapsed tumor remains a challenge.
CONFLICT OF INTEREST
None declared.
REFERENCES
- Kalapurakal JA, Dome JS, Perlman EJ, Malogolowkin M, Haase GM, Grundy P, et al.: Management of Wilms’ tumour: current practice and future goals. Lancet Oncol. 2004; 5: 37-46.
- Pritchard-Jones K, Kelsey A, Vujanic G, Imeson J, Hutton C, Mitchell C, et al.: Older age is an adverse prognostic factor in stage I, favorable histology Wilms’ tumor treated with vincristine monochemotherapy: a study by the United Kingdom Children’s Cancer Study Group, Wilm’s Tumor Working Group. J Clin Oncol. 2003; 21: 3269-75.
- D’Angio GJ, Breslow N, Beckwith JB, Evans A, Baum H, deLorimier A, et al.: Treatment of Wilms’ tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1989; 64: 349-60.
- de Kraker J, Graf N, van Tinteren H, Pein F, Sandstedt B, Godzinski J, et al.: Reduction of postoperative chemotherapy in children with stage I intermediate-risk and anaplastic Wilms’ tumour (SIOP 93-01 trial): a randomised controlled trial. Lancet. 2004; 364: 1229-35.
- Grundy P, Breslow N, Green DM, Sharples K, Evans A, D’Angio GJ: Prognostic factors for children with recurrent Wilms’ tumor: results from the Second and Third National Wilms’ Tumor Study. J Clin Oncol. 1989; 7: 638-47.
- Dome JS, Liu T, Krasin M, Lott L, Shearer P, Daw NC, et al.: Improved survival for patients with recurrent Wilms tumor: the experience at St. Jude Children’s Research Hospital. J Pediatr Hematol Oncol. 2002; 24: 192-8.
- Weirich A, Ludwig R, Graf N, Abel U, Leuschner I, Vujanic GM, et al.: Survival in nephroblastoma treated according to the trial and study SIOP-9/GPOH with respect to relapse and morbidity. Ann Oncol. 2004; 15: 808-20.
- Garaventa A, Hartmann O, Bernard JL, Zucker JM, Pardo N, Castel V, et al.: Autologous bone marrow transplantation for pediatric Wilms’ tumor: the experience of the European Bone Marrow Transplantation Solid Tumor Registry. Med Pediatr Oncol. 1994; 22: 11-4.
- Pein F, Michon J, Valteau-Couanet D, Quintana E, Frappaz D, Vannier JP, et al.: High-dose melphalan, etoposide, and carboplatin followed by autologous stem-cell rescue in pediatric high-risk recurrent Wilms’ tumor: a French Society of Pediatric Oncology study. J Clin Oncol. 1998; 16: 3295-301.
- Campbell AD, Cohn SL, Reynolds M, Seshadri R, Morgan E, Geissler G, et al.: Treatment of relapsed Wilms’ tumor with high-dose therapy and autologous hematopoietic stem-cell rescue: the experience at Children’s Memorial Hospital. J Clin Oncol. 2004; 22: 2885-90.
- Bonadio JF, Storer B, Norkool P, Farewell VT, Beckwith JB, D’Angio GJ: Anaplastic Wilms’ tumor: clinical and pathologic studies. J Clin Oncol. 1985; 3: 513-20.
- de Camargo B, Franco EL: A randomized clinical trial of single-dose versus fractionated-dose dactinomycin in the treatment of Wilms’ tumor. Results after extended follow-up. Brazilian Wilms’ Tumor Study Group. Cancer. 1994; 73: 3081-6.
- Green DM, Breslow NE, Beckwith JB, Finklestein JZ, Grundy P, Thomas PR, et al.: Effect of duration of treatment on treatment outcome and cost of treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1998; 16: 3744-51.
- Pianezza ML, Rubin S, Bass J, Chou S, Pike JG, Leonard MP: Wilms’ tumor at the Children’s Hospital of Eastern Ontario: 1990-2001. Can J Urol. 2004; 11: 2151-6.
- Green DM, Breslow NE, Beckwith JB, Ritchey ML, Shamberger RC, Haase GM, et al.: Treatment with nephrectomy only for small, stage I/favorable histology Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2001; 19: 3719-24.
- Grundy P, Perlman E, Rosen NS, Warwick AB, Bender JG, Ehrlich P, et al.: Current issues in Wilms tumor management. Curr Probl Cancer. 2005; 29: 221-60.
- Ehrlich PF, Ritchey ML, Hamilton TE, Haase GM, Ou S, Breslow N, et al.: Quality assessment for Wilms’ tumor: a report from the National Wilms’ Tumor Study-5. J Pediatr Surg. 2005; 40: 208-12.
- Seseke F, Zoller G, Ringert RH: Wilms’ tumor—single-center experience with renal surgery. Scand J Urol Nephrol. 2004; 38: 373-7.
- Ritchey ML, Shamberger RC, Haase G, Horwitz J, Bergemann T, Breslow NE: Surgical complications after primary nephrectomy for Wilms’ tumor: report from the National Wilms’ Tumor Study Group. J Am Coll Surg. 2001; 192: 63-8.
____________________
Accepted after revision:
October 15, 2006
_______________________
Correspondence address:
Dr. Silvio Tucci Jr
Hospital das Clínicas de Ribeirão Preto
University of São Paulo
Av. Bandeirantes, 3900
Ribeirão Preto, SP, 14048-900, Brazil
E-mail: stucci@convex.com.br
The
management of Wilms’ Tumor (WT) over the last two decades has seen
vast improvements in overall survival due to better neoadjuvant and adjuvant
therapeutic protocols developed by national and international collaborations
(1). With the survival improvements, came the need to reduce treatment
intensity so that the health burden on patients, parents and the health
care system was reduced without compromising outcome. The Brazilian collaborative
group (Grupo Cooperativo Brasileiro para o Tratamento do Tumor de Wilms
[GCBTTW]) (2) contributed to this international consensus by showing that
a single day administration of actinomycin-D rather than a 5-day course
(provided other treatment regimens were constant) was equally effective.
Tucci
et al., in this article, provide a retrospective summary of 53 children
treated at a single centre over a 14-year period. Although the retrospective
nature of the study is recognized, the authors should be congratulated
on providing further evidence that stage-I WT need not aggressive chemotherapeutic
regimens. Almost all of these patients did not have unfavorable histology
and thus the reduction in chemotherapy seems to be the correct regimen
for them. Such results are supported by the European and British groups,
both of whom have shown reduction of chemotherapy does not necessarily
lead to poorer outcome. The UK group showed that overall survival was
in the high nineties when vincristine was given only over ten weeks. However,
this applied to only those 4 years of age or younger (3). The SIOP have
reduced the pre-operative chemotherapy to 4 weeks and shown it is as good
as 8 weeks with no change of stage distribution and tumor shrinkage. Tucci
et al., in the present paper, mention the recent NWTSG-5 study showing
that of 75 patients younger than 2 years with a stage-I, favorable histology
WT less than 550g in weight treated without adjuvant chemotherapy 8 patients
developed recurrence to the lung or operative bed and 3 developed metachronous
contralateral WT. This resulted in a 2-year disease-free survival estimate
of 86.5%. Based on predefined stopping rules, this arm of the study was
closed early. However, subsequent review of these patients revealed several
factors that were not considered in these predefined stopping rules, the
most important being that the overall survival rate of these patients
was much higher than estimated. This suggested that, even if these children
relapse, the ability to successfully control the relapse was far greater
than predicted. Based on this, the newly formed US Children’s Oncology
Group will again evaluate this question in this group (4).
The
outcome for relapsed WT is worrying with intensive treatment having variable
effect. Certainly, this cohort seems to have worse outcome compared to
that reported elsewhere, but with the patients recruited over a period
of 14 years, many of whom were treated before more recent studies, there
is likely to be heterogeneity in management and possibly even histological
evaluation. The authors may have given consideration to a review of all
histology by expert histopathologists and application of recent staging
criteria so that such effects could be evaluated. The future for these
patients may be in high-dose chemotherapy with autologous stem cell transplant,
but as exemplified by the one patient they treated the toxicity of treatment
can be distressing for all concerned. Recent molecular studies have started
to characterize genetic changes that may stratify relapses into high and
low risk (5). Such advances could identify subgroups that may have higher
risk of failure after attempted salvage with intensive chemotherapeutic
regimens. In such patients, the risk: benefit ratio may be more appealing
when considering autologous stem-cell transplant and chemotherapy.
REFERENCES
- Ahmed HU, Arya M, Tsiouris A, Sellaturay SV, Shergill IS, Duffy PG, et al.: Update on the management of Wilms’ tumour. Eur J Surg Oncol. 2007; Feb 19, [Epub ahead of print].
- de Camargo B, Franco EL: A randomized clinical trial of single-dose versus fractionated-dose dactinomycin in the treatment of Wilms’ tumor. Results after extended follow-up. Brazilian Wilms’ Tumor Study Group. Cancer. 1994; 73: 3081-6.
- Mitchell C, Morris-Jones P, Kelsey A, Vujanic GM, Marsden B, Shannon R, et al.: The treatment of Wilms’ tumour: results of the United Kingdom children’s cancer study group (UKCCSG) second Wilms’ tumour study. Br J Cancer. 2000; 83 :602–8.
- Perlman EJ: Pediatric renal tumors: practical updates for the pathologist. Pediatr Dev Pathol. 2005;8: 320-38.
- Natrajan R, Little SE, Sodha N, Reis-Filho JS, Mackay A, Fenwick K, et al.: Analysis by array CGH of genomic changes associated with the progression or relapse of Wilms’ tumour. J Pathol. 2007; 211: 52-9.
Dr. Hashim U Ahmed
Dr. Manit Arya
Dr. Imran Mushtaq
Great Ormond Street Hospital for Children
The Institute of Urology & Nephrology
Division of Surgical and Interventional Sciences
University College London, UK
E-mail: hashim.ahmed@ucl.ac.uk
EDITORIAL COMMENT
I
read with great interest the article titled “Results of Novel Strategies
for the Treatment of Wilms’ Tumor” and I would like to congratulate
Silvio Tucci Jr et al. The article represents a critical, in-depth contribution
to the issue of contemporary Wilms’ tumor treatment.
Wilms’ tumor, represents approximately 6% of all childhood cancers
and is the most common primary malignant renal tumor of childhood. Current
management emphasizes in reducing the morbidity of treatment for low-risk
patients and reserving more intensive treatment for selected high-risk
patients for whom survival remains poor.
There
is a well known debate going on according to the treatment protocol one
must use. The International Society of Pediatric Oncology (SIOP) Protocols
have always recommended preoperative chemotherapy because it is able to
reduce tumor size, induce a pseudocapsule and decrease the incidence of
tumor rupture (1). Indeed, the authors describe an above average tumor
rupture in their study. The National Wilms’ Tumor Study Group (NWTSG),
on the other hand, recommends preoperative chemotherapy in some cases
only; bilateral tumors, inoperable tumors at surgical exploration and
inferior vena cava extension above the hepatic veins. This allows precise
staging of patients with modulation of treatment for each individual,
thereby decreasing the intensity of treatment toxicity. The authors have
chosen the NWTSG protocol, while in Europe the SIOP protocol is more popular
and therefore mostly used.
I
would also like to congratulate the author’s surgical approach.
I believe that transperitoneal radical nephrectomy with regional lymph
node sampling is the optimal surgical approach for Wilms’ tumor
patients, since it allows complete inspection of abdominal cavity, lymph
node sampling and tumor resection with lower percentage rates of neoplastic
cell spillage (2).
Reduced
postoperative chemotherapy seems to be a good solution in stage I patients
since their cure rate is not compromised. High dose chemotherapy with
autologous stem-cell rescue in children with relapsed Wilms’ tumor
exhibits variable disease free survival rates and needs to be further
studied since it is not clear if it offers any advantages over conventional
second line therapies. Treatment intensification for children with high-risk
tumors, although accompanied with sever complications, seems to be at
the time the only solution for such patients.
REFERENCES
- Graf N, Tournade MF, de Kraker J: The role of preoperative chemotherapy in the management of Wilms tumor. The SIOP studies. International Society of Pediatric Oncology. Urol Clin North Am. 2000; 27: 443-54.
- Zugor V, Krot D, Schott GE: Risk factors for intra- and postoperative complications in Wilms’ tumor surgery. Urologe A. 2007; 46: 274-7.
Dr. Vahudin Zugor
Department of Urology
Friedrich-Alexander-Universitat
Erlangen-Nurnberg
Erlangen, Germany
E-mail: vahudin.zugor@uro.imed.uni-erlangen.de