INDICATIONS FOR
GENETIC EVALUATION OF MEN
IN A REPRODUCTIVE MEDICINE PROGRAM
DAVID M. NUDELL, RODRIGO PAGANI, LARRY I. LIPSHULTZ
Division
of Male Reproductive Medicine and Surgery, Scott Department of Urology,
Baylor College of Medicine, Houston, Texas, USA
ABSTRACT
The technique of in vitro fertilization (IVF) with intracytoplasmic sperm injection (ICSI) has become widespread for the treatment of severe forms of male factor infertility. Although ICSI has allowed many men previously felt to be poor candidates for IVF the chance for biologic paternity, there have been concerns raised regarding possible transmission of abnormal genes to offspring using this technique. In particular, infertile men with severe defects in sperm production have been shown to have a significantly higher rate of genetic abnormalities than fertile men (1-3). IVF with ICSI clearly bypasses critical natural selection barriers that normally might prevent genetic disease transmission. For this reason, without long-term follow up and evaluation of children born using this technique, there is now a greater need for genetic evaluation in some couples undergoing IVF with ICSI. The reasons for evaluating possible genetic abnormalities include the determination of a cause of the infertility, and the risk of genetic propagation of disease. We now have a clearer understanding of how known, established genetic defects such as cystic fibrosis transmembrane regulator (CFTR) gene mutations and Klinefelter syndrome, for example, affect infertile couples and their offspring. The last several years, however, have led to several advances in genetic detection such as the delineation of Y chromosome abnormalities and the identification of increasingly common chromosome structural abnormalities in men with severe infertility. For men who remain currently untreatable even with ICSI, there may soon be the potential for fertilization with more primitive cells in the spermatogenic pathway such as round spermatids. The genetic implications of this new genetic vector remain unknown. In addition, we may soon be capable of repopulating testes devoid of germ cells with stem cells capable of undergoing complete spermatogenesis. This review examines recent genetic advances in the area of male infertility and summarizes current indications and testing available for genetic evaluation in infertile men.
Key words:
male infertility; reproduction; genetics; azoospermia; microdelections
Braz J Urol, 27: 105-119, 2001
INTRODUCTION
A
male factor is currently diagnosed in nearly 50% of infertile couples.
Despite this, roughly half of these men continue to have an unknown or
idiopathic cause for their subfertility. Recently, however, genetic advances
have made it possible to classify many disorders previously considered
idiopathic. This has led to the understanding that men with severe forms
of infertility may harbor genetic defects as the primary cause of their
infertility. Prior to ICSI, with natural intercourse, intrauterine insemination
(IUI) or even standard IVF, only the best sperm were capable of penetrating
the egg, thus leading to fertilization. It has been known for some time
that poor sperm morphology, for example, leads to significantly reduced
success with standard IVF (4). With ICSI, these types of natural selection
barriers are lost. Indeed, the “selection” of sperm for ICSI
involves the mere microscopic appearance of an intact sperm chosen by
the ICSI technician. This type of selection may be concerning in that
as many as 19% of sperm from normal men have chromosome structural abnormalities
when tested with the sensitive technique of fluorescent in situ hybridization
(FISH) (5). Similarly, men with severe oligospermia (< 10 million sperm/ml),
but who have plentiful sperm to undergo ICSI, are at risk for harboring
Y-chromosome deletions that have been shown to be passed to male offspring
(6). Men born with congenital bilateral absence of the vas deferens (CBAVD)
often have cystic fibrosis gene mutations despite exhibiting no other
sequelae of the disease. As more primitive spermatogenic cells become
feasible for use with ICSI, there will no doubt be further concerns for
the possibility of propagating genetic defects. Thus, genetic testing
and counseling is becoming increasingly important in men with severe infertility.
One diagnostic tool that may become more
useful for severely infertile men undergoing ICSI is preimplantation genetic
diagnosis (PGD). PGD allows for genetic evaluation of embryos derived
from ICSI procedures prior to their reimplantation. This technique is
accomplished through genetic evaluation of a single cell taken as a microscopic
biopsy from an early 8-cell embryo. Single cell analysis can then be done
either with standard karyotyping, FISH, or the polymerase chain reaction
(PCR). PGD can be used to detect chromosome numerical and structural disorders,
Y-chromosome deletions and CF gene mutations. Other disease states such
as XYY syndrome, Tay-Sachs disease, fragile X syndrome and Duchenne’s
muscular dystrophy can also be evaluated (7). As our ability to detect
genetic abnormalities improves using molecular techniques, this will be
an increasingly common procedure done in conjunction with ICSI.
Because the technology of IVF continues
to rapidly evolve, it is critical that clinicians offer and understand
genetic testing available to the couples whom they are treating. Despite
known genetic risks, there remains no consensus from organizations such
as the American Society of Reproductive Medicine, American College of
Obstetrics and Gynecology and the American Urologic Association regarding
specific indications for genetic testing in couples undergoing ICSI. It
is generally accepted, however, that men with either azoospermia or severe
oligospermia (< 5 million sperm/ml) are at the greatest risk of harboring
genetic defects and thus should undergo testing. In addition, those in
whom a suspicion for genetic disease exists based on family history, phenotype,
or past IVF outcomes, should also undergo tailored genetic testing.
The goal of this review is to examine our
clinical approach to the diagnosis of genetic abnormalities in infertile
men. The focus will be on specific genetic conditions that can be diagnosed
during the infertility evaluation. For simplicity, genetic disorders will
be categorized as classic Mendelian, chromosomal numerical or chromosomal
structural disorders. Subsequently, newer methods of genetic testing including
preimplantation genetic diagnosis (PGD) will be discussed. Finally, specific
indications for genetic testing based on the patient’s presenting
characteristics will be presented.
MENDELIAN DISORDERS
Mendelian
disorders (Table-1) are caused by a mutation at a single genetic locus.
These defects can occur de novo or can be inherited through autosomal
dominant, autosomal recessive or X-linked patterns (3). Mendelian disorders
commonly observed in infertile men are described below.

Cystic
Fibrosis Transmembrane
Gene Mutations (1:2500)
Cystic Fibrosis (CF) is the most common
autosomal recessive disease in caucasians with an incidence of 1:2500
births and a carrier frequency of 1:20 (3). The cystic fibrosis transmembrane
regulator gene (CFTR; 7q31.2) was cloned in 1989 and encodes a cyclic
adenosine monophosphate-regulated chloride channel found in many secretory
epithelia (8). To date, over 800 mutations have been identified in the
CFTR gene (9).
Clinical features of CF include chronic
pulmonary infection, exocrine pancreatic insufficiency, neonatal meconium
ileus and male infertility. Over 95% of men have abnormalities in Wolffian
Duct derived structures manifested most commonly as congenital bilateral
absence of the vas deferens (CBAVD) (1,2). Anatomically, the body and
tail of the epididymis, vas deferens, seminal vesicles and ejaculatory
ducts are affected, but the testicular efferent ducts and the caput epididymis
are invariably present due to the non-Wolffian derived nature of these
structures. Spermatogenesis is usually but not necessarily normal in affected
men (10). Because of the potentially fatal and incurable nature of the
disease, both men with CBAVD and their partners should undergo CF testing.
About 2% of infertile men not suffering
from cystic fibrosis are found to have CBAVD (11). The CFTR gene contains
27 exons and is 250 base pairs in length. A three base pair deletion in
exon 10 (delta F508) accounts for 70% of the mutations found in the caucasian
population. Of note is that considerable variation exists amongst different
racial groups (3). Fifty to 80% of men with CBAVD and 43% with congenital
unilateral absence of the vas (CUAVD) have detectable CFTR gene mutations
(12,13). Interestingly, Mickle et al. showed that those patients with
CUAVD and CFTR gene mutations often had a non-iatrogenic contralateral
vasal occlusion located superior to the scrotum (14). CFTR gene mutations
have also been found in men with CBAVD but without other manifestations
of CF (15). It is now understood that a majority of these men are compound
heterozygotes for different mutations in each allele of the CFTR gene.
A common mutation consists of a DNA variant in a non-coding sequence,
the 5T (thymidines) allele in the 3’ splicing region of intron 8
(15). Seven or nine thymidines usually occur in this region; a reduction
to the 5-thymidine variant decreases the efficiency of splicing of exon
9 and eventually leads to a 10-50% reduction in CFTR mRNA (3). This reduction
in mRNA appears to lead to CBAVD without the systemic features of CF.
The risk of CFTR gene mutations in patients with nonobstructive azoospermia
(NOA) remains unclear. While some studies have shown these mutations to
exist in patients with idiopathic testis failure, others have not shown
CFTR gene mutations in these patients (16). We continue to recommend that
all patients with CBAVD, CUAVD or idiopathic obstruction be screened for
CFTR gene mutations.
Generalized Wolffian duct anomalies can
also occur in patients with CBAVD. These include partial epididymal aplasia
and seminal vesicle aplasia or hypoplasia, which may lead to low ejaculate
volume. Secondary findings can include ipsilateral renal agenesis in 11%
with CBAVD and in 26% with unilateral vassal absence (13). Imaging confirmation
of renal agenesis is imperative in patients with unilateral absence of
the vas deferens or in those with CBAVD lacking detectable CFTR gene mutations.
Kallmann
Syndrome (1:30000)
Kallmann syndrome, or idiopathic hypogonadotropic
hypogonadism, is inherited as a familial disorder in one third of cases.
Both X-linked and autosomal inheritance patterns have been described (1).
In the X-linked recessive form, deletions occur in kalig-1 (kallmann-interval
1 gene), a gene responsible for the migration of GnRH neurons to the preoptic
area of the hypothalamus during development (17). As a consequence, there
is failure of testicular stimulation by the anterior pituitary and resultant
testis failure. In recent animal work, defects in the GnRH receptor itself
have been observed that suggest the mechanism for a second, autosomal
inheritance pattern (18).
The clinical manifestations of Kallmann
syndrome depend on the degree of hypogonadism. Most patients experience
a delay in puberty although those with less severe defects may present
with a normal appearing phenotype and only subfertility. Other findings
include anosmia, cleft palate and small testes. Testicular biopsies can
demonstrate a wide range of findings from focal areas of complete spermatogenesis
to germ cell aplasia (9). Medical treatment is highly successful and consists
of gonadotropin replacement over a 12-18 month period, which induces sperm
in the ejaculate in 80% of affected men (19). Often, gonadotropin replacement
and eventual stimulation of spermatogenesis can be accomplished with human
chorionic gonadotropin alone. In cases where this fails, the addition
of human menopausal gonadotropin is useful.
Androgen
Receptor Gene Mutations (1:60000)
Over 300 mutations have been found in the
androgen receptor, a large steroid receptor gene located on the X chromosome
(Xq11-q12) (1). In addition to well-recognized mutations within the gene’s
8 exons, mutations in the gene promoter region have also been reported
(3). Because many mutations exist, the syndrome is clinically variable
and ranges from phenotypic females (complete androgen insensitivity, testicular
feminization) to normally virilized but infertile males (20). Depending
on the severity of the defect, serum testosterone levels can be low, normal
or high. The androgen concentration in each individual depends on the
functional integrity of the androgen receptors within the pituitary and
hypothalamus.
Recent genetic research on the androgen
receptor gene has also led to interesting new clinical correlation with
male infertility. The androgen receptor gene has 8 exons and it is known
that a critical region of CAG-nucleotide repeats, usually 15-30 in number,
can be found in exon 1 (4). Elongation of this repeat region results in
spinal and bulbar muscular atrophy (Kennedy disease), a neurodegenerative
disorder that begins around age 30 and consists of muscle cramping and
atrophy as well as infertility from testicular atrophy. There is now evidence
that subtle abnormalities in this CAG repeat region may also underlie
some cases of idiopathic infertility. Yoshida et al. recently detected
longer than normal CAG nucleotide repeats in normally virilized men with
normal genitalia and idiopathic azoospermia (21). These added CAG nucleotides
may be the result of a failure to recognize and repair DNA abnormalities
in dividing cells. Whether this represents a more global DNA repair enzyme
problem is currently unknown. This does, however, suggest that defects
in the androgen receptor may underlie more of male infertility than previously
recognized.
Immotile
Cilia Syndrome (1:30000)
The hallmark of this heterogeneous disease
is impaired or absent ciliary and/or flagellar motility. Clinical manifestations
include chronic cough and sinus infection, nasal polyposis, bronchiectasis,
and infertility. Kartagener’s syndrome, a particular form of this
disease (1:60000) consists of situs inversus, bronchiectasis, chronic
sinusitis and infertility. Immotile cilia syndromes are characterized
by abnormal cell axonemes, both in cilia and sperm tails. The normal axonemal
arrangement is 9 outer microtubular doublets with inner and outer dynein
arms surrounding 2 central microtubules. In this disease, various defects
in either the microtubule and/or dynein arm assembly have been reported
(22). It is thought that flagellar function is controlled by the action
of over 200 distinct genes (3). Although no single gene to date has been
found, the inheritance pattern derived from family pedigrees suggests
that transmission is likely to be autosomal recessive.
While infertility is universal in patients
with immotile cilia syndromes, ejaculated sperm can be motile and sperm
concentrations can be normal or even high. With ICSI, clinical pregnancies
and live births have now been reported from men with this syndrome (22,23).
The use of unselected, immotile sperm from such men carries normal fertilization
rates of 20 to 40% (24). However, with the use of techniques like hypo-osmotic
swelling to select viable, nonmotile sperm, fertilization is possible
(25). Because the gene defect is usually recessive, normal offspring are
likely in these cases; still, genetic counseling is critical. Interestingly,
a novel association between immotile ciliary syndromes and autosomal dominant
polycystic kidney (ADPK) disease has recently been observed in Japanese
patients: 25% of men with immotile cilia syndromes in this study also
had ADPK (26). The pathophysiological relationship between these two disorders
is currently unknown.
Autosomal
Dominant Polycystic Kidney Disease (ADPK)(1:400-1:1000)
Numerous large cysts of the kidneys, liver,
pancreas and spleen characterize this autosomal dominant disorder, and
a 10-40% chance of developing berry aneurysms in the brain. Because the
syndrome is often asymptomatic until adulthood, affected men may initially
present with infertility. Cysts in the epididymis, seminal vesicle or
ejaculatory ducts that compress the ductal system and cause obstruction
(26) cause infertility. Three separate genetic loci have been associated
with ADPK. PKD1 accounts for 85% frequency of the disease and is found
on chromosome 16p13.3, PKD2 has been mapped to chromosome 4q, and PKD3
is currently unmapped (3,27). Azoospermic ADPK patients can produce offspring
with ICSI using sperm harvested from the epididymis or testis, necessitating
appropriate genetic counseling.
CHROMOSOMAL DISORDERS AND MALE INFERTILITY
Chromosomal
disorders (Table-2) are defined as the loss, gain or abnormal arrangement
of genetic material at the chromosomal level. These disorders can be further
divided into numerical and structural abnormalities. Among numerical abnormalities,
polyploidy is defined as a chromosome number that is an exact multiple
of 23 (excluding 46) (2). Aneuploidy is the gain or loss of one or more
chromosomes, such as in Klinefelter syndrome (47,XXY) or Down syndrome
(trisomy 21). These defects can occur in all cells or only in some cells,
a condition termed “mosaicism”. Structural chromosome disorders
can occur in single or multiple chromosomes and are becoming more frequently
recognized as contributing factors to infertility. Examples of defects
that occur in a single chromosome are deletions, duplications and inversions.
Translocations, either balanced or unbalanced, occur between 2 chromosomes.
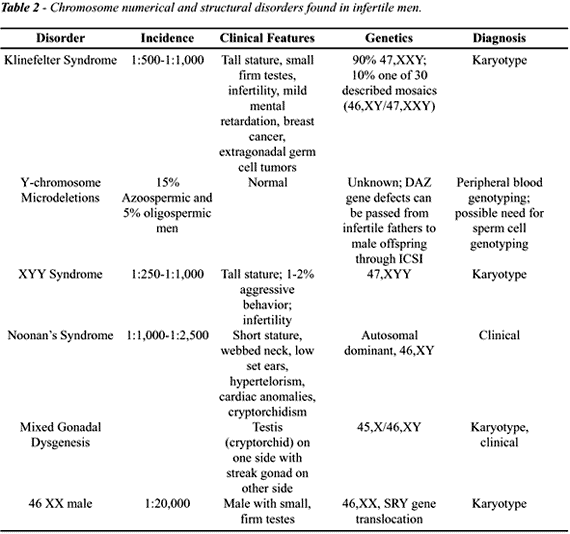
Chromosomal disorders are found much more
frequently in the infertile men than in fertile individuals (28). These
chromosome disorders currently can be detected in 15% of azoospermic and
5% of oligospermic men, and now represent one of the most common genetic
defects in infertile men (29,30). Recognition of these defects will continue
to improve as our ability to detect them improves. With ICSI, infertile
patients with these disorders that were previously considered untreatable
can now be offered paternity (28). As such, it is important that these
couples undergo genetic testing.
Klinefelter
Syndrome (1:500-1:1000)
Klinefelter Syndrome (47,XXY) is 45 times
more common in men seeking treatment for infertility than in the general
male population and is the most common numerical chromosome anomaly seen
in male infertility (28). About 90% of men have the classic 47,XXY genotype;
the remainders have any number of 30 recognized mosaic patterns (1). Clinically,
fewer than 5% of patients present before puberty. Most men present in
adulthood with tall stature, gynecomastia (25%) and infertility. Affected
men are also predisposed to diabetes mellitus, varicose veins, chronic
bronchitis, extragonadal germ cell tumors and breast tumors. Among infertile
men, the classic phenotype is a patient with small, firm testes, elevated
serum FSH, and low serum testosterone and elevated estradiol levels. Patients
are commonly azoospermic and testis biopsy often reveals hyalinization
of the seminiferous tubules with Leydig cell hyperplasia. However, there
may be focal areas of spermatogenesis (9). The mosaic variant is less
severe and can present with normal testicular size, complete spermatogenesis
and the presence of ejaculated sperm.
Pregnancies in infertile men with Klinefelter
syndrome have now been reported using ICSI (31). For this reason, there
has been recent interest in determining germ cell ploidy (number of sex
chromosomes) in sperm obtained from these patients to assess whether sperm
are normal haploid (X or Y chromosome present) or hyperhaploid (XX, XY
etc. present). In one study, the risk of non-disjunction and hyperhaploid
sperm production as determined by 3-color FISH was estimated to be 3.9%
of all sperm (32). With the use of preimplantation genetic diagnosis (PGD),
it is now possible to detect abnormal aneuploid embryos and avoid their
reimplantation (33).
47,XYY
Syndrome (1:250 - 1:1000)
The genetic cause of this syndrome is paternal
non-disjunction during meiosis that results in YY sperm (3). These patients
exhibit tall stature, and 1-2% show aggressive behavioral characteristics.
Most are azoospermic or severely oligospermic and testis biopsy findings
range from Sertoli cell-only to maturation arrest patterns (9). Unlike
Klinefelter syndrome, serum testosterone levels are normal.
Noonan’s
Syndrome (1:1000-1:2500)
Features of this clinical syndrome are similar
to Turner syndrome in females and include short stature, webbed neck,
hypertelorism, low set ears, cubitus valgus, ptosis, and cardiovascular
anomalies. While an X-linked dominant pattern of inheritance was originally
suspected, it is now known that at least one gene defect is inherited
in an autosomal dominant manner on chromosome 12 (1). Patients typically
have hypogonadism with low testosterone and high pituitary gonadotropins.
Cryptorchidism and testicular atrophy are commonly observed in this condition.
Mixed
Gonadal Dysgenesis
Patients with mixed gonadal dysgenesis usually
have a mosaic 45,XO/46,XY genotype and anatomically have a testis on one
side and a streak gonad on the other (1). Often, the normally formed testis
is intraabdominal and devoid of viable germ cells. The streak gonad is
at risk of developing gonadoblastoma or seminoma and should be surgically
removed. There are varying degrees of ambiguity of the external genitalia.
Translocations
and Inversions
An increasing number of men with previously
diagnosed idiopathic infertility are found to harbor chromosomal translocations
and inversions as our ability to detect these defects improves. In men
with very low sperm counts, subtotal structural abnormalities can now
be observed in 2 to 4% despite a lack of detectable aneuploidy (30). In
these men, the rate of balanced translocations and gene inversions is
8 times that of normal men (30). Roughly half of these defects are Robertsonian
translocations between chromosome 13 and 14. Robertsonian translocations
occur when the short arm of 2 acrocentric chromosomes (chromosomes with
very little genetic material on the short arm) fuses. This rearrangement
gives rises to a single chromosome containing the long arms of the two
chromosomes while the short arms are lost. The translocation is considered
balanced despite the loss of one chromosome due to the paucity of genetic
information present on the lost short arm. These patients typically have
severe spermatogenic defects but can produce viable embryos with ICSI
(28). However, unbalanced translocations can occur in the offspring. Although
male infertility due to translocations is potentially treatable with preimplantation
genetic diagnosis, few viable pregnancies have been achieved to date.
Y chromosome
Microdeletions
Tiepolo & Zuffardi were first to show
that the Y chromosome (Yq11) (Figure-1), might contain an azoospermia
factor (AZF) (34). Reijo et al., almost 20 years later, discovered specific
microdeletions in the Y chromosome in azoospermic men (Figure-2) (35).
At that time, one candidate gene region for AZF was thought to be RBM
(RNA Recognition Motif); however, these authors demonstrated that a separate
region of Yq was deleted in 13% of azoospermic men and termed this gene
DAZ (Deleted in Azoospermia) (35). Subsequently, Vogt et al. described
deletions in 3 distinct regions of the Y chromosome, which they termed
Azoospermia Factor regions a, b and c (AZFa, b, c) (36). The DAZ gene
cluster is found in AZFc (Figure-2). Kent-First et al. have recently suggested
the presence of a fourth AZF region (AZFd) residing between AZFb and AZFc
that may be associated with mild impairment of spermatogenesis and abnormal
sperm morphology (37). Importantly, it is now clear that similar Yq microdeletions
also occur in 4 to 8% of severely oligospermic men (38). Taken together,
these deletions represent the most common molecularly defined cause of
male infertility in humans (39).

Reproductive center to center variation
in detection rates of Y chromosome microdeletions is large and is most
likely due more to non-standardization of detection techniques than natural
differences in populations (40). Another complicating feature of Y chromosome
analysis is that some men may harbor DAZ deletions only in germ line tissue
(mosaics) and not in somatic cells (6). Thus, the current technique of
testing peripheral blood with standard PCR techniques may not be truly
reflective of an individual’s Y chromosome status in germ-line tissue.
The actual status of a patient’s Y chromosome for infertility purposes
may require analysis of individual sperm cells to define the incidence
of this possible mosaicism.

The relationship between AZFa, b and c deletions
and the histology of the testis in infertile men remains undefined. In
general, variable testis histology can accompany any AZF deletion and
ICSI may be potentially successful in all groups (39,41). However, it
has been shown conclusively that genetic deletions in the DAZ region can
be passed to the male offspring of affected fathers through ICSI (42).
Thus, 1 in 10,000 live births could be affected by DAZ deletions if one
assumes 1 in 1000 men is azoospermic (2).
The DAZ gene encodes an RNA-binding protein
thought to be critical for spermatogenesis (35). During evolution, the
DAZ gene complex was most likely transposed to the Y chromosome since
many other organisms have autosomal homologues to the DAZ gene (so called
DAZ-Like (DAZL)). Recently, an autosomal homologue to human DAZ has been
found on chromosome 3 but its function is unknown. The DAZL genes in other
species have strong functional relationships to fertility. Homozygous
DAZ-deleted mice exhibit sterility whereas heterozygous mice are only
subfertile. Disruption of the DAZL homologue boule in flies results in
maturation arrest and sterility. Such variation in the degree of infertility
with deletion status suggests that the copy number of normal DAZ/DAZL
may influence the eventual fate of male germ cells (38). In humans, the
DAZ protein product is present in high concentration in premeiotic germ
cells (43), corroborating findings from other studies that localize DAZ
mRNA to similar cell populations in the mouse (44).
Meiosis
and DNA Repair
Maturation arrest is commonly observed in
testis biopsies from infertile men with nonobstructive azoospermia. The
morphological point of arrest (primary spermatocyte) is suggestive of
a defect in meiosis, since secondary spermatocytes have already undergone
crossing-over of DNA, the critical step whereby DNA is exchanged between
the maternal and paternal alleles. Recently, many genes required to complete
meiosis in other organisms have been identified (45). Some of these genes
appear to tightly regulate the recombination of genetic material and the
repair of obligate DNA breakages that are essential for recombination
to occur during meiosis. In organisms such as yeast and mice, mutations
in such genes required for DNA repair lead invariably to infertility characterized
by meiotic arrest (possible equivalent to human maturation arrest) (46).
There is literature to suggest that defects
in DNA repair may also underlie human infertility. Pearson et al. observed
a reduced ability to repair induced DNA damage in lymphocytes from an
azoospermic man with arrested germ cell growth (47). Solari et al. recently
found abnormal meiotic synaptonemal complexes in testis biopsies from
infertile men (48). These observations suggest that human maturation arrest
may indeed be genetic and may be linked to mutations in genes required
for DNA repair.
Recently, using microsatellite DNA analysis
to evaluate DNA repair fidelity, we have found that the ability to repair
DNA mismatches is altered in some infertile patients with maturation arrest
(49,50). In these men, DNA repair defects occurred only in the germ cells
(dividing by meiosis) and not in somatic cells (dividing by mitosis) where
DNA recombination does not occur (50). Some individuals with incomplete
maturation arrest (sperm present focally) also were found to harbor similar
defects in DNA repair as men with complete arrest. Some of these men will
have enough sperm present for ICSI and thus could pass DNA repair defects
to their offspring. At this time, however, the relationship of defective
DNA repair in germ cells and the subsequent health of children conceived
by ICSI is not known.
NEW DIRECTIONS IN GENETICS
Spermatid
Injection
Several reports have shown that fertilization
and cleavage of embryos can be achieved with both elongated and round
spermatids, potentially offering paternity to men with late maturation
arrest (51,52). Fertilization rates approaching that seen with fully mature
sperm (50 to 71%) have been observed with elongated spermatids (ELSI)
and several live births have been reported (51). However, the fertilization
rates with round spermatid injection (ROSI) are still quite disappointing
(20 to 25%) (51,53). Despite the poor efficiency of the technique, there
have been reports of live births from ROSI (52). Whether these births
occurred with the use of round spermatids or elongated spermatids remains
controversial (54). The question of whether round spermatids have undergone
all of the critical genetic maturation processes necessary for normal
development remains unanswered.
Germ Cell
Transplantation
The concept of transplanting early spermatogenic
cells offers the hope of germ cell repopulation in sterile testicles and
forms a basis for research in totipotent stem cells (stem cell renewal)
(55). Recently, studies have shown that rat spermatagonial cells that
are transplanted into the seminiferous tubules of immunodeficient mice
can generate rat spermatogenesis (56). In addition, it may now be possible
to cryopreserve spermatagonial stem cells (56) and propagate them in culture
to encourage their development and maturation into later spermatogenic
forms (57). Clearly, this would be useful in men undergoing chemotherapy
or radiation therapy for malignancies. Research in this field in the next
decade has tremendous promise to enrich the field of male infertility
from both scientific and clinical viewpoints.
Preimplatation
Genetic Diagnosis (PGD)
The importance of early detection of genetic
disorders in the offspring of couples treated with ICSI is critical. The
first embryo biopsy was performed in 1990 and used the polymerase chain
reaction (PCR) to detect Y-chromosome bearing sperm in order to avoid
implantation of embryos with an X-linked disorder (58). As of 1998, normal
embryos have been identified and replaced with the help of PGD from patients
with genetic diseases ranging from Mendelian gene disorders to those with
numerical or structural chromosomal defects (33). Most couples undergo
PGD for one of 3 reasons: a)- genetic risk with previous spontaneous abortion,
b)- genetic risk with objection to therapeutic abortion, and c)- genetic
risk with subfertility (7). Embryo biopsy success rate (97%) and the accuracy
of PGD in diagnosing specific abnormalities have been very high in general,
with only one known misdiagnosis as of 1997 (33). However, because of
the complexity and invasive nature of the technique, the overall pregnancy
rate for PGD embryos is lower than those observed with standard ICSI (17.6%
vs. 25-33%).
PGD has been used to diagnose many of the
genetic conditions described in this chapter. For CF gene mutations, Handyside
et al reported the first use of PGD to detect the delta F508 mutation
(59). It has now been shown that several other CF gene mutations can be
rapidly evaluated from a single cell PGD biopsy (60). Although technically
feasible, the use of PGD for Y-chromosome deletions has not been described.
The may be due to the lack of a noticeable disease state in the affected
offspring. Perhaps most importantly, PGD has been used to detect chromosomal
aneuploidies using multiprobe FISH (61). This is most useful in patients
with mosaic Klinefelter syndrome at risk of propagating sex chromosome
aneuploidy or those with advanced maternal age at risk of Down syndrome.
Finally, PGD has been used to detect reciprocal translocations in chromosomes
5 and 8 (64) as well as Robertsonian translocations (63). The detection
of Robertsonian translocations may be important because it has recently
been shown that significant pregnancy rates are achieved despite the presence
of this defect (64).
CONCLUSIONS
As our ability to bypass the detrimental effects of genetic defects on fertility improves, it is imperative that physicians treating infertile couples understand and continue to examine genetic causes of infertility. There have been tremendous advances in our understanding of some diseases and their relationship to infertility such as that for CFTR gene mutations and CBAVD. We now appear similarly close to elucidating molecular steps in sperm production with our evolving understanding of genes on the Y chromosome and genes involved with meiosis. A greater mechanistic understanding of normal spermatogenesis will not only help explain the cause of infertility in many men, but also the risk these men face as ART techniques become increasingly more widespread. With genetic counseling, family genetic analysis, and evolving PGD technology, many of the genetic risks that severely infertile men bring to ICSI can be potentially avoided in the future.
REFERENCES
- Bhasin S, Ma K, Sinha I, Limbo M, Taylor WE, Salehian B: The genetic basis of male infertility. Endocrinol Metab Clin North Am, 27: 783-805, 1998.
- Kupker W, Schwinger E, Hiort O, Ludwig M, Nikolettos N, Schlegel PN, Diedrich K: Genetics of male subfertility: consequences for the clinical work-up. Hum Reprod, 14: 24-37, 1999.
- Mak V, Jarvi KA: The genetics of male infertility. J Urol, 156: 1245-1256, 1996.
- Kruger TF, Acosta AA, Simmons KF, Swanson RJ, Matta JF, Oehninger S: Predictive value of abnormal sperm morphology in in-vitro fertilization. Fertil Steril, 49: 112-117, 1988.
- Bischoff FZ, Nguyen DD, Burt KJ, Shaffer LG: Estimates of aneuploidy using multicolor fluorescence in situ hybridization on human sperm. Cytogenet Cell Genet, 66: 237-243, 1994.
- Kent-First MG, Kol S, Muallem A, Ofir R, Manor D, Blazer S, First N, Itskovitz-Eldor J: The incidence and possible relevance of Y-linked microdeletions in babies born after intracytoplasmic sperm injection and their infertile fathers. Mol Hum Reprod, 2: 943-950, 1996.
- Kim ED, Bischoff FZ, Lipshultz LI, Lamb DJ: Genetic concerns for the subfertile male in the era of ICSI. Prenat Diagn, 18: 1349-1365, 1998.
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC: Identification of the cystic fibrosis gene: genetic analysis. Science, 245: 1073-1080, 1989.
- Patrizio P, Broomfield D: The Genetic Basis of Male Infertility. In: Glover TD, Barrat CLR (ed.), Male Fertility and Infertility. Cambridge, Cambridge University Press, 162-179, 1999.
- Meng M, Turek PJ: Impaired spermatogenesis in men with congenital absence of the vas deferens. Fertil Steril, 72: 177, 1999.
- Jequier AM: Obstructive azoospermia: a study of 102 patients. Clin Reprod Fertil, 3: 21-36, 1985.
- Patrizio P, Asch RH, Handelin B, Silber SJ: Aetiology of congenital absence of vas deferens: genetic study of three generations. Hum Reprod, 8: 215-220, 1993.
- Schlegel PN, Shin D, Goldstein M: Urogenital anomalies in men with congenital absence of the vas deferens. J Urol, 155: 1644-1648, 1996.
- Mickle J, Milunsky A, Amos JA, Oates RD: Congenital unilateral absence of the vas deferens: a heterogeneous disorder with two distinct subpopulations based upon aetiology and mutational status of the cystic fibrosis gene. Hum Reprod, 10: 1728-1735, 1995.
- Jarvi K, Zielenski J, Wilschanski M, Durie P, Buckspan M, Tullis E, Markiewicz D, Tsui LC: Cystic fibrosis transmembrane conductance regulator and obstructive azoospermia [letter]. Lancet, 345: 1578, 1995.
- Mak V, Zielenski J, Tsui LC, Durie P, Zini A, Martin S, Longley TB, Jarvi KA: Cystic fibrosis gene mutations and infertile men with primary testicular failure. Hum Reprod, 15: 436-439, 2000.
- Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, Carrozzo R, Maestrini E, Pieretti M, Taillon-Miller P: A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature, 353: 529-536, 1991.
- Layman LC, Cohen DP, Jin M, Xie J, Li Z, Reindollar RH, Bolbolan S, Bick DP, Sherins RR, Duck LW, Musgrove LC, Sellers JC, Neill JD: Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism [letter]. Nat Genet, 18: 14-15, 1998.
- Buchter D, Behre HM, Kliesch S, Nieschlag E: Pulsatile GnRH or human chorionic gonadotropin/human menopausal gonadotropin as effective treatment for men with hypogonadotropic hypogonadism: a review of 42 cases. Eur J Endocrinol, 139: 298-303, 1998.
- Shkolny DL, Beitel LK, Ginsberg J, Pekeles G, Arbour L, Pinsky L, Trifiro MA: Discordant measures of androgen-binding kinetics in two mutant androgen receptors causing mild or partial androgen insensitivity, respectively. J Clin Endocrinol Metab, 84: 805-810, 1999.
- Yoshida KI, Yano M, Chiba K, Honda M, Kitahara S: CAG repeat length in the androgen receptor gene is enhanced in patients with idiopathic azoospermia. Urology, 54: 1078-1081, 1999.
- Von Zumbusch A, Fiedler K, Mayerhofer A, Jessberger B, Ring J, Vogt HJ: Birth of healthy children after intracytoplasmic sperm injection in two couples with male Kartagener’s syndrome. Fertil Steril, 70: 643-646, 1998.
- Nijs M, Vanderzwalmen P, Vandamme B, Segal-Bertin G, Lejeune B, Segal L, Van Roosendaal E, Schoysman R: Fertilizing ability of immotile spermatozoa after intracytoplasmic sperm injection. Hum Reprod, 11: 2180-2185, 1996.
- Barros A, Sousa M, Oliveira C, Silva J, Almeida V, Beires J: Pregnancy and birth after intracytoplasmic sperm injection with totally immotile sperm recovered from the ejaculate. Fertil Steril, 67: 1091-1094, 1997.
- Smikle CB, Turek PJ: Hypo-osmotic swelling can accurately assess the viability of nonmotile sperm. Mol Reprod Dev, 47: 200-203, 1997.
- Okada H, Fujioka H, Tatsumi N, Fujisawa M, Gohji K, Arakawa S, Kato H, Kobayashi S, Isojima S, Kamidono S: Assisted reproduction for infertile patients with 9 + 0 immotile spermatozoa associated with autosomal dominant polycystic kidney disease. Hum Reprod, 14: 110-113, 1999.
- Kimberling WJ, Kumar S, Gabow PA, Kenyon JB, Connolly CJ, Somlo S: Autosomal dominant polycystic kidney disease: localization of the second gene to chromosome 4q13-q23. Genomics, 18: 467-472, 1993.
- Van Assche E, Bonduelle M, Tournaye H, Joris H, Verheyen G, Devroey P, Van Steirteghem A, Liebaers I: Cytogenetics of infertile men. Hum Reprod, 11: 1-24; 1996.
- Pandiyan N, Jequier AM: Mitotic chromosomal anomalies among 1210 infertile men. Hum Reprod, 11: 2604-2608, 1996.
- Peschka B, Leygraaf J, Van der Ven K, Montag M, Schartmann B, Schubert R, Van der Ven H, Schwanitz G: Type and frequency of chromosome aberrations in 781 couples undergoing intracytoplasmic sperm injection. Hum Reprod, 14: 2257-2263, 1999.
- Palermo GD, Schlegel PN, Sills ES, Veeck LL, Zaninovic N, Menendez S, Rosenwaks Z: Births after intracytoplasmic injection of sperm obtained by testicular extraction from men with nonmosaic Klinefelter’s syndrome. N Engl J Med, 338: 588-590, 1998.
- Bielanska M, Tan SL, Ao A: Fluorescence in-situ hybridization of sex chromosomes in spermatozoa and spare preimplantation embryos of a Klinefelter 46,XY/47,XXY male. Hum Reprod, 15: 440-444, 2000.
- Geraedts J, Handyside A, Harper J, Liebaers I, Sermon K, Staessen C, Thornhill A, Vanderfaeillie A, Viville S: ESHRE Preimplantation Genetic Diagnosis (PGD) Consortium: preliminary assessment of data from January 1997 to September 1998. ESHRE PGD Consortium Steering Committee. Hum Reprod, 14: 3138-3148, 1999.
- Tiepolo L, Zuffardi O: Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet, 34: 119-124, 1976.
- Reijo R, Lee TY, Salo P, Alagappan R, Brown LG, Rosenberg M, Rozen S, Jaffe T, Straus D, Hovatta O: Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet, 10: 383-393, 1995.
- Vogt PH, Edelmann A, Kirsch S, Henegariu O, Hirschmann P, Kiesewetter F, Kohn FM, Schill WB, Farah S, Ramos C, Hartmann M, Hartschuh W, Meschede D, Behre HM, Castel A, Nieschlag E, Weidner W, Grone HJ, Jung A, Engel W, Haidl G: Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet, 5: 933-943, 1996.
- Kent-First M, Muallem A, Shultz J, Pryor J, Roberts K, Nolten W, Meisner L, Chandley A, Gouchy G, Jorgensen L, Havighurst T, Grosch J: Defining regions of the Y-chromosome responsible for male infertility and identification of a fourth AZF region (AZFd) by Y-chromosome microdeletion detection. Mol Reprod Dev, 53: 27-41, 1999.
- Reijo R, Alagappan RK, Patrizio P, Page DC: Severe oligozoospermia resulting from deletions of azoospermia factor gene on Y chromosome. Lancet, 347: 1290-1293, 1996.
- Kostiner DR, Turek PJ, Reijo RA: Male infertility: analysis of the markers and genes on the human Y chromosome. Hum Reprod, 13: 3032-3038, 1998.
- Kleiman SE, Yogev L, Gamzu R, Hauser R, Botchan A, Lessing JB, Paz G, Yavetz H: Genetic evaluation of infertile men. Hum Reprod, 14: 33-38, 1999.
- Kremer JA, Tuerlings JH, Meuleman EJ, Schoute F, Mariman E, Smeets DF, Hoefsloot LH, Braat DD, Merkus HM: Microdeletions of the Y chromosome and intracytoplasmic sperm injection: from gene to clinic. Hum Reprod, 12: 687-691, 1997.
- Mulhall JP, Reijo R, Alagappan R, Brown L, Page D, Carson R, Oates RD: Azoospermic men with deletion of the DAZ gene cluster are capable of completing spermatogenesis: fertilization, normal embryonic development and pregnancy occur when retrieved testicular spermatozoa are used for intracytoplasmic sperm injection. Hum Reprod, 12: 503-508, 1997.
- Nudell D, Dorfman D, Reijo R, Turek P: The expression of DAZ and DAZL proteins in human testes with normal and abnormal spermatogenesis. J Urol, 159: A881, 1998.
- Niederberger C, Agulnik AI, Cho Y, Lamb D, Bishop CE: In situ hybridization shows that Dazla expression in mouse testis is restricted to premeiotic stages IV-VI of spermatogenesis. Mamm Genome, 8: 277-278, 1999.
- Roeder GS: Meiotic chromosomes: it takes two to tango. Genes Dev, 11: 2600-2621, 1997.
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A, Ashley T, Liskay RM: Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet, 13: 336-342, 1996.
- Pearson PL, Ellis JD, Evans HJ: A gross reduction in chiasma formation during meiotic prophase and a defective DNA repair mechanism associated with a case of human male infertility. Cytogenetics, 9: 460-467, 1970.
- Solari AJ: Synaptonemal complex analysis in human male infertility. Eur J Histochem, 43: 265-276, 1999.
- Kim ED, Levy N, DeLara J, Lipshultz LI, Lamb, DJ: Genomic instability is increased in states of abnormal spermatogenesis in male infertility. J Urol, 161: A1197, 1999.
- Nudell D, Castillo M, Turek PJ, Reijo-Pera R: Defective DNA repair in infertile men. Hum Reprod, 15:1289-1294, 2000.
- Al-Hasani S, Ludwig M, Palermo I, Kupker W, Sandmann J, Johannisson R, Fornara P, Sturm R, Bals-Pratsch M, Bauer O, Diedrich K: Intracytoplasmic injection of round and elongated spermatids from azoospermic patients: results and review. Hum Reprod, 14: 97-107, 1999.
- Kahraman S, Polat G, Samli M, Sozen E, Ozgun OD, Dirican K, Ozbicer T: Multiple pregnancies obtained by testicular spermatid injection in combination with intracytoplasmic sperm injection. Hum Reprod, 13: 104-110, 1998.
- Ghazzawi IM, Alhasani S, Taher M, Souso S: Reproductive capacity of round spermatids compared with mature spermatozoa in a population of azoospermic men. Hum Reprod, 14: 736-740, 1999.
- Silber SJ, Johnson L, Verheyen G, Van Steirteghem A: Round spermatid injection [editorial]. Fertil Steril, 73: 897-900, 2000.
- Brinster RL, Zimmermann JW: Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci U S A, 91:11298-11302, 1994.
- Nagano M, Brinster RL: Spermatogonial transplantation and reconstitution of donor cell spermatogenesis in recipient mice. Apmis, 106: 47-55, 1998.
- Tesarik J, Greco E, Rienzi L, Ubaldi F,Guido M, Cohen-Bacrie P, Mendoza C: Differentiation of spermatogenic cells during in-vitro culture of testicular biopsy samples from patients with obstructive azoospermia: effect of recombinant follicle stimulating hormone. Hum Reprod, 13: 2772-2781, 1998.
- Handyside AH, Kontogianni EH, Hardy K, Winston RM: Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature, 344: 768-770, 1990.
- Handyside AH, Lesko JG, Tarin JJ, Winston RM, Hughes MR: Birth of a normal girl after in vitro fertilization and preimplantation diagnostic testing for cystic fibrosis. N Engl J Med, 327: 905-909, 1992.
- Scobie G, Woodroffe B, Fishel S, Kalsheker N: Identification of the five most common cystic fibrosis mutations in single cells using a rapid and specific differential amplification system. Mol Hum Reprod, 2: 203-207, 1996.
- Munne S, Sultan KM, Weier HU, Grifo JA, Cohen J, Rosenwaks Z: Assessment of numeric abnormalities of X, Y, 18, and 16 chromosomes in preimplantation human embryos before transfer. Am J Obstet Gynecol, 172: 1191-1199, 1995.
- Pierce KE, Fitzgerald LM, Seibel MM, Zilberstein M: Preimplantation genetic diagnosis of chromosome balance in embryos from a patient with a balanced reciprocal translocation. Mol Hum Reprod, 4: 167-172, 1998.
- Conn CM, Harper JC, Winston RM, Delhanty JD: Infertile couples with Robertsonian translocations: preimplantation genetic analysis of embryos reveals chaotic cleavage divisions. Hum Genet, 102: 117-123, 1998.
- Munne
S, Sandalinas M, Escudero T, Fung J, Gianaroli L, Cohen J: Outcome of
preimplantation genetic diagnosis of translocations. Fertil Steril,
73: 1209-1218, 2000.
________________________
Received: December 5, 2000
Accepted: December 22, 2000
_______________________
Correspondence address:
Dr. Larry I. Lipshultz
Male Reproductive Medicine and Surgery
Scott Department of Urology
6560 Fannin Suite 2100
Houston, TX 77030, USA
Fax: + + (1) (713) 798-6007
E-mail: larryl@bcm.tmc.edu