PRIMARY
RENAL SARCOMA WITH MORPHOLOGIC AND IMMUNOHISTOCHEMICAL ASPECTS COMPATIBLE
WITH SYNOVIAL SARCOMA
(
Download pdf )
CARLOS H. SCHAAL, FÁBIO C. NAVARRO, FRANCISCO A. MORAES NETO
Amaral Carvalho Hospital, Jaú, São Paulo, Brazil
ABSTRACT
Primary synovial sarcoma of the kidney is a rare tumor that is difficult to diagnose. We present one case that was not diagnosed through fine needle aspiration, requiring a morphologic and immunohistochemical analysis of the incision biopsy. Since the tumor was surgically unresectable, chemotherapy was employed previously to definitive radical surgery.
Key
words: kidney neoplasms; synovial sarcoma; surgery; chemotherapy
Int Braz J Urol. 2004; 30: 210-3
INTRODUCTION
The synovial sarcoma is an uncommon tumor, representing approximately 6 to 10% of the primary sarcomas of soft tissues. Recently, rare primary cases were described in the kidney (1-3). Signs and symptoms are similar to any primary renal tumor. Unspecific preoperative findings, associated with its rarity, prevent its early diagnosis.
CASE REPORT
Male,
27-year old patient, presenting gross hematuria and large abdominal mass.
The abdominal computed tomography showed a large retroperitoneal mass,
radiologically consistent with malignant neoplasia (Figure-1). Aspiration
puncture of the lesion was performed with fine needle, diagnosing small
cell malignant neoplasia (Figure-2).

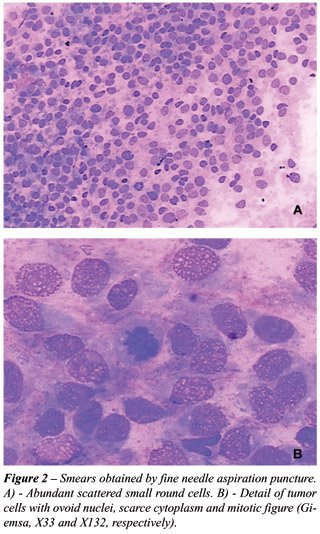
During staging, no distant lesions were
observed. Due to the large tumor volume, an option was made to performing
adjuvant chemotherapy. A chemotherapy protocol for Wilms’ tumor
was started, with actinomycin 50 mg/Kg and vincristine 0.04 mg/Kg, for
4 cycles, obtaining minimal response, with the mass remaining unresectable.
Open biopsy of the lesion was indicated,
aiming a better subtyping of the neoplasia. Histopathological examination
revealed a malignant neoplasia constituted by 2 distinct cell populations,
one consisting of small cells with round hyperchromatic nuclei and high
mitotic index (poorly differentiated subtype with high malignancy grade)
and the other of spindle cells with oval nuclei, fine chromatin, indistinct
nucleoli and low mitotic index (monophasic fibrous subtype with low malignancy
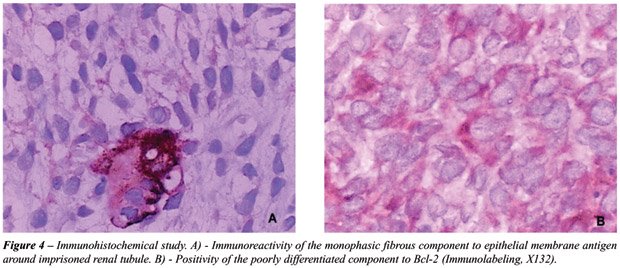
grade) surrounding renal tubules cystically dilated (Figure-3). The immunohistochemical
study was consistent with synovial sarcoma: positive for pan-cytokeratin,
epithelial membrane antigen, vimentin, Bcl-2 and negative for CD99/MIC-2,
muscle-specific desmin and actin HHF-35 (Figure-4). A new chemotherapy
cycle was promptly started with iphosphamide 50 mg/Kg and adriamycin 1
mg/Kg, for 4 weeks.

With the considerable reduction of the tumor
(approximately 50%), radical nephrectomy by right thoracolaparotomy was
indicated, with partial exeresis of diaphragm and ascending colon being
necessary. No macroscopically involved lymph nodes were found intraoperatively.
Histopathological examination of the surgical
specimen revealed an encapsulated tumoral mass measuring 21 x 15 x 9 cm,
weighting 1628 g, with extensive areas of necrosis and hemorrhage. The
adrenal gland, ureter or renal vein were not involved, and the lymph node
analysis was negative for neoplasia. Patient has been followed for 1 year
with no signs of recurrence.
DISCUSSION
Primary
synovial sarcoma of the kidney is a type of renal sarcoma. Leiomyosarcoma
represents 40 - 60% of the described renal sarcomas, followed by rhabdomyosarcoma,
histiosarcoma, chondrosarcoma and osteosarcoma, liposarcoma, angiosarcoma
and hemangiopericytoma (1-3). Only 20 cases were reported in the literature.
It affects young individuals, of both genders, between 20 and 50 years,
presenting a clinical picture that is similar to renal tumors in general
(1). There is no clinical or imaging characteristic that can indicate
the diagnosis (1-5). Differential diagnosis with other renal tumors constituted
by spindle or round cells is difficult, requiring immunohistochemical
studies or molecular analysis. Among the differential diagnoses, are especially
the mesoblastic nephroma and adult neuroectodermal tumor (PNET), in addition
to adult Wilms’ tumor and fibrosarcoma.
The mesoblastic nephroma is usually immunoreactive
for actin and negative for epithelial markers. The synovial sarcoma presents
immunoreaction for epithelial markers such as cytokeratin and epithelial
membrane antigen, generally with focal distribution. The proteins CD99
and Bcl-2 were detected in 70% and 100% of cases, respectively. PNET tumors
are immunoreactive to CD99 and generally negative for Bcl-2 and epithelial
membrane antigen. However, since there is not a specific immunohistochemical
marker for the synovial sarcoma, definitive diagnosis can be achieved
only by identifying the characteristic T chromosomal translocation (X;
18), through reverse transcriptase polymerase chain reaction with fluorescence
in situ hybridization (FISH) (1-3). In the present case, since we could
not count on chromosomal analysis, diagnosis was established based on
conventional morphologic microscopic analysis and immunohistochemistry.
In one research on 17 cases (1), 4 presented
lung, liver or bone metastases and evolved to death, 2 presented pelvic
recurrence.
Adjuvant and neoadjuvant chemotherapy, using
high doses of doxorubicin, cisplatin and iphosphamide (14 g/m2) performed
in 14 patients without initial metastatic disease, rendered 93% of patients
disease-free in a mean follow-up of 37 months (6 to 85 months) (5).
The base for chemotherapy in such cases
is the iphosphamide, usually reducing the tumor volume by 50% or more
(3,5). There is no consensus in the literature concerning the standard
chemotherapy scheme. In this case, the scheme consisting in iphosphamide
and adriamycin reduced approximately 50% of the tumor, enabling its surgical
resection.
REFERENCES
- Argani P, Faria PA, Epstein JI, Reuter VE, Pearlman EJ, Beckwith JB, et al.: Primary renal synovial sarcoma. Am J Surg Pathol. 2000; 24: 1087-96.
- Bella AJ, Winquist EW, Pearlman EJ: Primary synovial sarcoma of the kidney diagnosed by molecular detection of SYT-SSX fusion transcripts. J Urol. 2002; 168: 1092-3.
- Koyama S, Morimitsu Y, Morokuma F, Hashimoto H: Primary synovial sarcoma of the kidney: report of a case confirmed by molecular detection of the SYT-SSX2 fusion transcripts. Pathol Int. 2001; 51: 385-91.
- Kim DH, Sohn JH, Lee Mc, Lee G, Yoon GS, Hashimoto H, et al.: Primary synovial sarcoma of the kidney. Am J Surg Pathol. 2000; 24: 1097-104.
- Kampe CE, Rosen G, Eilber F, Eckardt J, Lowenbraun S, Foster J, et al.: Synovial sarcoma. A study of intensive chemotherapy in 14 patients with localized disease. Cancer. 1993; 72: 2161-9.
_______________________
Received:
October 2, 2003
Accepted after revision: January 9, 2004
_______________________
Correspondence address:
Dr. Carlos Hermann Schaal
Rua Luís Paiva, 100
Jaú, SP, 17210-180, Brazil
Fax: + 55 14 3624-5155
E-mail: schaal.jau@uol.com.br