COLLECTING
DUCT CARCINOMA ASSOCIATED WITH ONCOCYTOMA
(
Download pdf )
GEORGE M. YOUSEF, GERSHON C. EJECKAM, LEONICO M. BEST, ELEFTHERIOS P. DIAMANDIS
Discipline of Pathology, Memorial University, St. John’s, Health Care Corporation of St. John’s, Newfoundland, and Department of Pathology and Laboratory Medicine, Mount Sinai Hospital, Toronto, Ontario, Canada
ABSTRACT
Collecting duct carcinoma (CDC) is a rare, highly aggressive malignant neoplasm that arises from the collecting duct epithelium of the kidney. CDC was reported to coexist with renal cell and transitional cell carcinomas. We report a rare case of CDC associated with oncocytoma, confirmed by the characteristic histological appearance and immunohistochemistry. We also review the epidemiological, histological and immunohistochemical criteria for diagnosis, in addition to the genetic and cytogenetic aberrations reported in the literature. Identification and reporting CDC is important for the establishment of treatment strategies and monitoring prognosis.
Key
words: kidney neoplasms; collecting ducts, kidney; oncocytoma
Int Braz J Urol. 2005; 31: 465-9
CASE REPORT
A 66-year old male with a history of non-insulin dependent diabetes, hypertension and chronic prostatitis presented gross hematuria, abdominal pain and renal failure. At the time of diagnosis, a CT scan showed a 4 cm poorly enhancing mass at the anterior aspect of the lower pole of the kidney, with associated lymph node and spinal cord metastasis. The patient underwent radical nephrectomy and was placed on hemodialysis until he died shortly following the procedure.
Pathologic
Findings
The main tumor mass was 6.0 x 5.0 x 5.0 cm in
the lower pole of the kidney. The cut surface was firm and grayish white
with focal areas of hemorrhage and necrosis. In addition, there were multiple
coalescing small grayish nodules scattered throughout the kidney.
Microscopically, the tumor consisted of a high-grade
tubulopapillary and solid lesion. Micropapillary to frank papillary patterns
with central fibrovascular stalks were present. Nuclei were large, vesicular
and possessed large prominent nucleoli. In some areas, the tumor showed
a tubulo-glandular pattern on a background of a desmoplastic stroma infiltrated
by inflammatory cells. Extensive tumor necrosis with calcification was
present. Tumor cells infiltrated the renal capsule, perirenal fat, and
the adrenal gland. Extensive lymphatic and vascular invasion were present.
On immunohistochemistry, tumor cells were positive to Ulex European agglutinin
1 lectin, vimentin, and distal tubular marker EMA (Figure-1). Another
nodule of a renal oncocytoma, measuring 1.0 cm in diameter with extensive
hemorrhage was also found in the renal cortex (Figure-2). This was positive
for AE1/AE3, negative for CK7 and Hale’s colloidal iron and shows
mitochondria on electron microscopy.
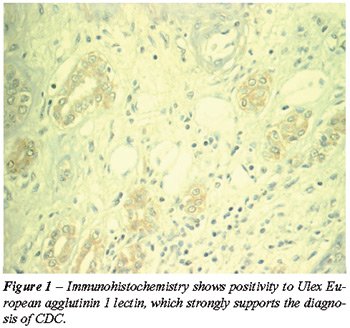
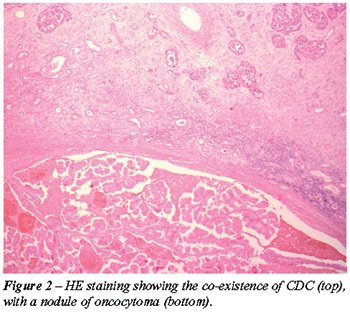
COMMENTS
Collecting duct carcinoma (CDC) is a rare, highly aggressive renal cell
carcinoma. On presentation, CDC is metastasized to regional lymph nodes
in about 80% of cases. There is a white male preponderance (1).
Grossly, the tumor may commence at the cortico-medullary
junction but due to its aggressiveness, can spread to the entire kidney
and beyond on diagnosis. Unlike conventional renal cell carcinoma, CDC
is not usually circumscribed, shows no bright yellow coloration and only
small to punctate hemmorrhagic areas. Microscopically, CDC is a high-grade
malignancy with large vesicular nuclei and large prominent nucleoli. There
is no accumulation of macrophages in the stroma. These features assist
in differentiating CDC from papillary renal cell carcinoma. On immunohistochemical
studies, tumor cell positivity with antibodies to Ulex European agglutinin
1 lectin strongly suggests the diagnosis of CDC. The tumor is also positive
to peanut agglutinin (PNA), vimentin, lysozyme, distal tubular marker
EMA, and high molecular weight cytokeratin, and negative for proximal
tubular markers (Leu-M1) (2). This is in contrast to conventional (clear
cell) renal cell carcinoma, which expresses pancytokeratin but is negative
for high molecular weight cytokeratin. CDC produces intracellular mucin,
which can be demonstrated by mucicarmine and PAS stains.
Chromosomal features of CDC are distinct. Comparative
genomic hybridization revealed a gain of chromosomal material on chromosome
3 and a loss of chromosomal material on chromosomes 12 and 22 (3). There
is also a loss of heterozygosity of 8p and 13q. A loss of chromosome arms
6p, 8p, 13q and 21q was reported in 40-50% of cases.
The coexistence of multiple primary cancers in
the kidney has been reported. CDC is reported to co-exist with other renal
tumors as RCC and transitional cell carcinoma. To our knowledge, this
is the first report that shows this rare coincidence of CDC with oncocytoma.
The association of CDC with oncocytoma is not surprising, since a similar
origin from the distal nephron was postulated for both.
Oncocytoma should be differentiated from the
closely related chromophobe renal cell carcinoma (CRCC). In CRCC, the
nuclei are wrinkled with hyperchromatic nuclei while those of oncocytoma
are perfectly round. Binucleation and multinucleation is more common in
CRCC. Well-defined cell membranes and perinuclear halos are seen in CRCC.
The cytoplasm Hale’s colloid iron staining is only positive in CRCC.
In electron microscopy, CRCC shows cytoplasmic vesicles while oncocytoma
shows mitochondria.
In conclusion, we report on a case of CDC which shows a rare association
with renal oncocytoma. Accumulation of data on CDC will allow for the
establishment of better treatment strategies and monitoring prognosis.
REFERENCES
- Storkel S, Eble JN, Adlakha K, Amin M, Blute ML, Bostwick DG, et al.: Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 1997; 80: 987-9.
- Singh I, Nabi G: Bellini duct carcinoma: review of diagnosis and management. Int Urol Nephrol. 2002; 34: 91-5.
- Meloni-Ehrig AM: Renal cancer: cytogenetic and molecular genetic aspects. Am J Med Genet. 2002; 115: 164-72.
________________________
Received: February 24, 2005
Accepted after revision: June 29, 2005
________________________
Correspondence address:
Dr. George M. Yousef
Health Sciences Center
300 Prince Philip Drive
St. John’s, NL, A1B 3V6
Ontario, Canada
Fax: + 1 709 777 8625
E-mail: gyousef@mtsinai.on.ca
EDITORIAL COMMENT
The case report presented on page 465, “Collecting Duct Carcinoma
Associated with Oncocytoma,” gives us an opportunity to comment
on the importance of classifying the renal epithelial neoplasms in adults.
The most recent WHO publication brings the classification shown in Table-1
(1). Such neoplasms have completely distinct macroscopic, microscopic,
genetic features and clinical behavior, and a proper histological classification
is fundamentally important when determining the prognosis and management
of these tumors.
Oncocytoma is a benign epithelial neoplasm. There
are no consistent reports of recurrence or metastases, and surgical treatment
is curative.

Among the malignant tumors, the collecting duct
(or Bellini’s) carcinomas are the most aggressive. They are fortunately
rare (< 1%), affect young individuals and only half of the cases present
metastatic disease. Interestingly, they respond poorly to immunotherapy,
the classical treatment for renal cell carcinomas, and appear to be responsive
to chemotherapy (2).
Clear cell carcinoma, the most common type (70%),
is the second most aggressive with a mean survival of 69% in 5 years (3).
Its cystic or multilocular variant shows a very good behavior, with 100%
of survival in 5 years (4). 75% of the time, its carcinogenesis route
is related to loss of the VHL gene, which controls the hypoxia-induced
factor (HIF-1) related to angiogenesis, pH control, glucose transport,
cell proliferation and migration (5). The importance of this knowledge
is the development of new treatment lines using inhibitors of tyrosine
kinase and of the RAS/RAF signaling pathway, which are the focus in several
clinical trials, with encouraging results (6).
The sarcomatoid carcinoma is not a histological
subtype, but a dedifferentiation of any CCR types. It represents a poor
prognosis factor by itself (7).
The spindle cell and mucinous tubular carcinomas
are typically low-grade tumors and show good clinical behavior (8). Other
tumors, whether equally rare or recently described, have a still unknown
behavior, and both urologists and pathologists must be alert in order
to recognize these new entities.
REFERENCES
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA: World Health Organization Classification of Tumors. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs, Lyon, IARC Press, 2004.
- Chao D, Zisman A, Pantuck AJ, Gitlitz BJ, Freedland SJ, Said JW, et al.: Collecting duct renal cell carcinoma: clinical study of rare tumor. J Urol. 2002; 167: 71-4.
- Cheville JC, Lohse CM, Zincke H, Weaver AL, Blutte ML: Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 2003; 27: 612-24.
- Corica FA, Iczkowski KA, Cheng L, Zincke H, Blutte ML, Wendel A, et al.: Cystic renal cell carcinoma is cured by resection: a study of 24 cases with long-term followup. J Urol. 1999; 161: 408-11.
- Linehan WM, Walther MM, Zbar B: The genetics basis of cancer of the kidney. J Urol. 2003; 170: 2163-72.
- Mancuso A, Sternberg CN: What’s new in the treatment of metastatic kidney cancer? BJU Int. 2005; 95: 1171-80.
- Dall’Oglio MF, Lieberknecht M, Gouveia V, Sant’Anna AC, Leite KR, Srougi M: Sarcomatoid differentiation in renal cell carcinoma: prognostic implications. Int Braz J Urol. 2005; 31: 10-16.
- Parwani AV, Husain AN, Epstein JI, Beckwith JB, Argani P: Low-grade myxoid renal epithelial neoplasms with distal nephron differentiation. Hum Pathol. 2001; 32: 506-12.
Dr.
Katia R Leite
Laboratory of Clinical and Molecular Pathology
University of Sao Paulo, USP
Sao Paulo, SP, Brazil
E-mail: katiaramos@uol.com.br
EDITORIAL COMMENT
Renal cell carcinoma is a tumor comprising several histologic subtypes,
the molecular hallmarks of which support their classification as distinct
entities. The most common form is clear cell or conventional renal carcinoma,
which has been associated with mutations in the von Hippel-Lindau gene
and loss of hetrozygosity at chromosome 3p. Next in frequency is papillary
renal cell carcinoma associated with trisomies of chromosomes 7 and 17
and loss of chromosome Y. Chromophobe renal cell carcinoma is associated
with numerous specific chromosomal losses. Chromosomal and genetic studies
of collecting duct carcinomas are limited. The most characteristic changes
of these aggressive tumors are deletions involving chromosomes 1,6,8,14,15,21,
and 22. Renal oncocytoma, a benign neoplasm, most often does not present
any chromosomal abnormalities.
Multicentricity is a relatively common finding
in papillary renal cell carcinomas. Renal clear cell carcinoma occurs
in 38 to 55 percent of patients with von Hippel-Lindau syndrome. Lesions
tend to be bilateral and multicentric, are often associated with cysts,
and occur at an earlier age than sporadic renal clear cell carcinoma.
Association of histologic subtypes is a rare
event in kidney tumors. The reported case on page 465, “Collecting
Duct Carcinoma Associated with Oncocytoma,” seems to be the first
report of an association of collecting duct carcinoma (a very aggressive
tumor) and oncocytoma (a benign tumor). Recently, an intriguing association
of renal tumors was reported. The Birt-Hogg-Dubé (BHD) syndrome
has been reported in association with renal tumors of a variety of histologic
types (1). BHD was originally described in 1977 as genodermatosis characterized
by autosomal dominantly inherited pale yellow-white dome-shaped papules
on the face, neck, and upper trunk (2). In addition to cutaneous lesions,
BHD confers an increased incidence of renal tumors, lung cysts, and spontaneous
pneumothorax, and have linked the BHD gene to chromosome presence of an
unusual hybrid form of renal tumors with elements of oncocytoma, chromophobe
renal cell carcinoma, and occasional areas reminiscent of renal clear
cell carcinoma.
REFERENCES
1. Pavlovich
CP, Walther MM, Eyler RA, Hewitt SM, Zbar B, Linehan WM, Merino MJ: Renal
tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol. 2002; 26: 1542-52.
2. Birt AR, Hogg GR, Dube WJ: Hereditary multiple fibrofolliculomas with
trichodiscomas and acrochordons. Arch Dermatol. 1977; 113: 1674-7.
Dr.
Athanase Billis
Full-Professor of Pathology
State University of Campinas, Unicamp
Campinas, São Paulo, Brazil
E-mail: athanase@fcm.unicamp.br